
CAPA Software: The Complete Guide
CAPA software is the digital system of record that takes a non-conformance from the moment it is raised — by an audit finding, customer complaint, supplier issue, or safety incident — through investigation, root-cause analysis, corrective and preventive action, and Verified Closure with proof of effectiveness. A modern CAPA platform replaces the spreadsheet-and-email workflow that quietly closes findings without ever proving the fix worked. It is the system that makes ISO 9001 Clause 10.2, IATF 16949 Clause 10.2.3, the FDA QMSR (21 CFR Part 820 as amended February 2026, incorporating ISO 13485 § 8.5.2 and § 8.5.3 by reference), and AS9100 Clause 10.2 defensible under audit. And it is the difference between a quality program that learns from its findings and one that repeats them, year after year.
Most CAPA workflows close prematurely. The action is performed, a signature is captured, the record is filed — and three months later the same finding recurs. We call the discipline that prevents this Verified Closure: treating “complete” and “effective” as two separate events, with evidence required at both. Verified Closure is Certainty-owned vocabulary. It is the standard we hold every CAPA in our platform against, and it is the test we recommend every quality and EHS leader apply to their existing program.
CAPA at a glance
| What it is | A flexible platform that runs the full closed-loop CAPA workflow — initiation, investigation, action plan, verification, closure, recurrence monitoring |
| Who uses it | QA Managers, EHS Managers, Compliance Directors, Plant Managers, Supplier Quality Engineers |
| Core capabilities | Multi-source initiation, 5-Why / Fishbone / 8D investigation, action plan, Verified Closure, recurrence reporting, audit trail |
| Standards covered | ISO 9001 Clause 10, IATF 16949 Clause 10.2.3 (problem solving / 8D), AS9100 Clause 10.2, FDA QMSR (21 CFR Part 820 as amended February 2026 — CAPA via ISO 13485 § 8.5.2 / § 8.5.3), ISO 45001 / OSHA incident CAPA |
| Brand wedge | Verified Closure — Certainty-owned discipline of separating “complete” from “effective” with proof of recurrence prevention |
| Outcome | Lower repeat-finding rate, faster customer audit prep, defensible regulatory evidence, audit findings that actually stay closed |
Table of contents
- What is CAPA software?
- Verified Closure — Certainty’s CAPA-forward standard
- Core CAPA capabilities
- CAPA in regulatory context
- Method integrations — 8D, 5-Why, Fishbone, Pareto
- How to choose CAPA software — buyer’s framework
- CAPA rollout roadmap (6 steps)
- Frequently Asked Questions (FAQs)
- Related resources

What is CAPA Software?
CAPA software (corrective and preventive action software) is the digital system of record that drives a non-conformance through investigation, root-cause analysis, corrective and preventive action, and Verified Closure with written proof of effectiveness. The platform works wherever the non-conformance is raised. It captures the trigger event: an audit finding, customer complaint, supplier non-conformance, safety incident, or internal observation. The platform assigns an owner with a due date and enforces a formal investigation method (5-Why, Fishbone, 8D). Both corrective and preventive actions are recorded: the corrective fixes the immediate issue, the preventive addresses the systemic cause. The record only closes when verification evidence shows that the action worked.
The acronym “CAPA” originated in the FDA’s Quality System Regulation (21 CFR Part 820.100) for medical device manufacturers, but the discipline now sits at the centre of every modern quality and safety management system. ISO 9001:2015 made the corrective-action requirement explicit (Clause 10.2). IATF 16949 layered on the 8D problem-solving expectation (Clause 10.2.3). AS9100 added aerospace-specific verification depth. ISO 45001 brought the same logic to occupational health and safety. Whatever the industry, the CAPA workflow is the closed loop that turns a finding into a learning.
CAPA vs NCR vs Root Cause Analysis — disambiguation
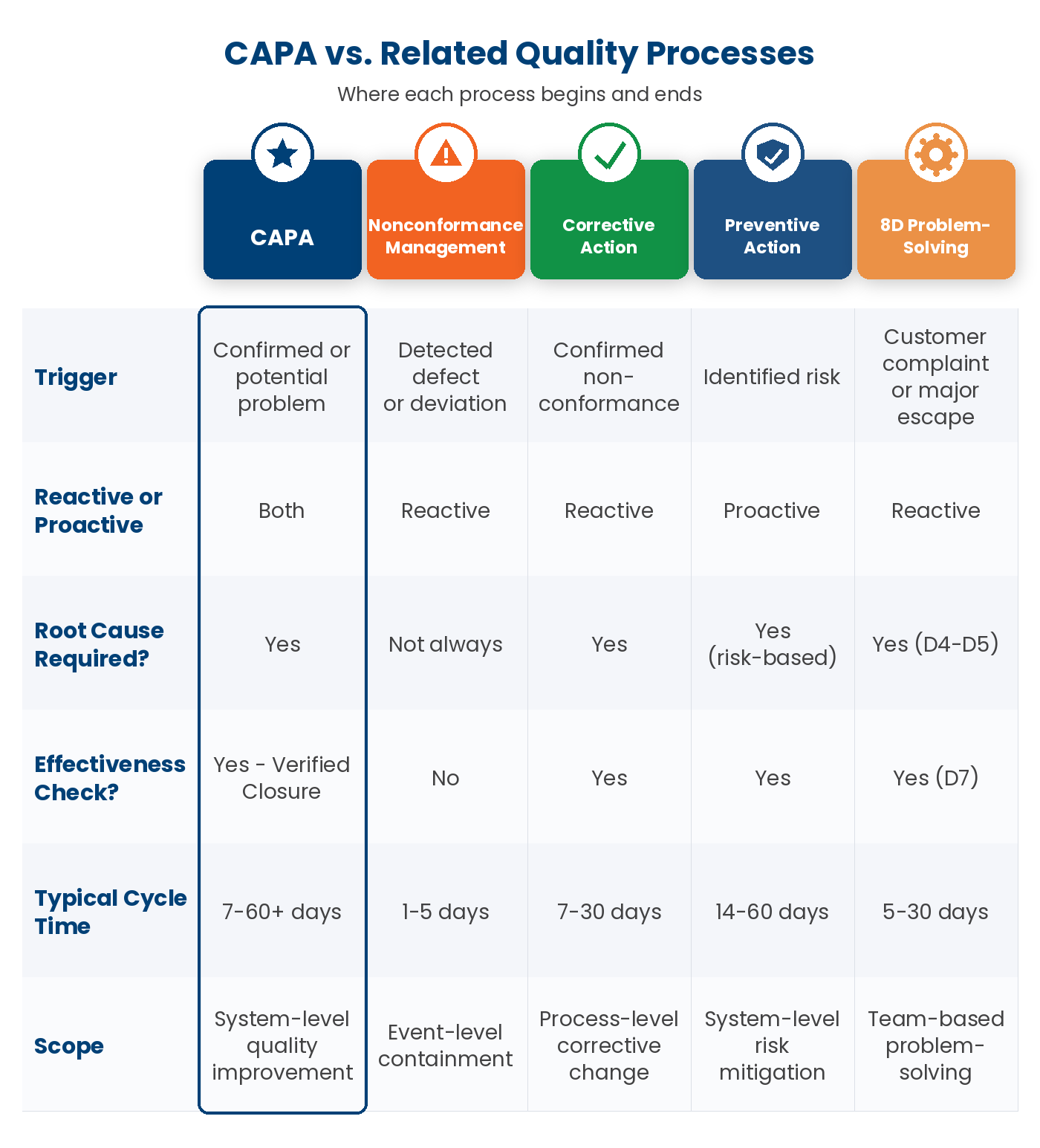
Three terms get conflated in CAPA conversations. They are related but distinct.
- Non-conformance (NCR). The documented deviation itself — a part out of spec, a procedure not followed, an audit finding raised, a customer complaint received. An NCR is the event. It is a single record that captures what happened, where, when, and against which specification or work instruction.
- Root Cause Analysis (RCA). The formal investigation that turns the NCR into a cause. RCA is the method — 5-Why, Fishbone, 8D, fault-tree, Pareto — used inside the CAPA workflow to determine why the deviation occurred. RCA is a step within CAPA, not a substitute for it.
- CAPA (Corrective and Preventive Action). The full closed-loop workflow. CAPA contains the NCR record and drives the RCA investigation. It captures the corrective action (immediate fix) and the preventive action (systemic fix to prevent recurrence). The record closes only after verification proves the action was effective.
In practice: an NCR without a CAPA is just a log entry. An RCA without a CAPA is an investigation report on a shelf. CAPA is the system that ties them together and keeps the record alive until recurrence is provably prevented.
Who uses CAPA software
CAPA software is bought and used across multiple ICPs because non-conformances arise everywhere — quality, safety, supplier, regulatory.
- QA Manager / Quality Manager. Owns the corrective-action workflow for product and process quality. Configures investigation templates, assigns owners, enforces Verified Closure, prepares for ISO 9001 and IATF 16949 surveillance audits.
- EHS Manager. Owns the CAPA workflow for safety incidents, near-misses, and ISO 45001 / OSHA recordable events. The same platform serves both quality and EHS in well-run organizations — one record-of-record, one verified-closure standard.
- Compliance Director. Owns the regulatory posture across CAPA-bearing standards (FDA cGMP, 21 CFR Part 820, IATF 16949, AS9100, ISO 45001). Reports up to executive leadership and out to certification bodies and regulators.
- Plant Manager / Operations Manager. Sees open CAPAs across their floor. Sponsors closure. Reviews recurrence rate as a daily operational metric, not a quarterly report.
- Supplier Quality Engineer. Issues supplier corrective-action requests (SCARs), manages supplier 8D responses, verifies effectiveness through follow-up inspection or supplier audit.
CAPA software is one of the few category buys that genuinely spans Quality, Safety, and Supplier Quality — which is why it tends to be the highest-leverage platform investment a manufacturer makes.
Industries served
CAPA discipline shows up wherever a defect, deviation, or incident has cost — financial, regulatory, or human. The largest concentrations are:
- Automotive and automotive supply — IATF 16949 mandates 8D problem solving (Clause 10.2.3); OEM customer-specific requirements add formal CAPA response timelines (often 24/48/72 hours for safety and major defects)
- Aerospace and defense — AS9100 demands rigorous root-cause discipline and verification depth for flight-safety parts
- Food and beverage — HACCP critical-control-point deviations route directly into CAPA with recall-risk implications
- Medical device — the FDA QMSR (21 CFR Part 820 as amended February 2026, incorporating ISO 13485:2016 by reference) makes CAPA a foundational FDA requirement; CAPA flows through ISO 13485 § 8.5.2 (corrective action) and § 8.5.3 (preventive action). CAPA records are inspected first in every FDA medical-device manufacturing inspection.
- Pharmaceutical and biotech — cGMP CAPA with 21 CFR Part 11 electronic-record requirements; CAPA closure cycle time is a regulator-watched metric
- Construction — incident CAPA tied to safety observations and near-miss programs; OSHA recordkeeping discipline
- Energy and utilities — process-safety CAPA under OSHA PSM, environmental incident CAPA under EPA reporting
The same flexible CAPA platform serves all of these — what changes is the trigger source, the method emphasis, and the closure-evidence requirement.
Verified Closure — Certainty’s CAPA-forward standard
Term: Verified Closure is the discipline of treating “action complete” and “action effective” as two separate events in a CAPA workflow. A corrective or preventive action is complete when the assigned owner has performed it and recorded the required evidence. A CAPA is closed only after a second, independent verification — a follow-up audit, a sample inspection, a fresh data point, or a recurrence-window observation — shows that the action actually prevented the original deviation from recurring.
Verified Closure is the single most important concept in this guide, and the reason the CAPA category exists as a brand wedge for Certainty. Most quality and EHS programs do not have a verified-closure step. They have an action-complete step that gets called “closure.” The result is written in every certification surveillance report and every plant manager’s frustration: the same findings, the same root causes, the same actions — year after year, audit after audit.
We treat Verified Closure as the defining test of a real CAPA system. A platform that does not enforce it — separately, with a different role signing off, with required evidence types, with a verification window — is a logging tool, not a CAPA tool. The same is true of a process: a CAPA SOP that allows the action owner to also be the verifier is a closure-on-signature program with a CAPA name.
Why most CAPA workflows close prematurely
Three patterns repeat across every organization we work with.
First, CAPA closure is signature-based, not evidence-based. The action owner ticks a box that says “complete,” uploads a single document, and the CAPA closes. Nothing in the workflow asks whether the action worked. No one routes the record back to the original auditor or the affected operator. The system never checks the same control point 30 or 60 days later for recurrence.
Second, the verifier is the same person as the action owner — or worse, the action owner’s direct report. Verification under these conditions is administrative, not investigative. The auditor who raised the finding never sees the closure; the quality engineer who performed the action signs their own work as effective. Independence collapses.
Third, the CAPA is severed from the original trigger. The audit finding lives in the audit module, the customer complaint in the CRM, the supplier issue in a supplier scorecard, and the CAPA in a third system. None of them talk to each other. When the same control point fails again six months later, no one connects the new finding to the closed CAPA — because the CAPA is in another database.
How a CAPA-forward platform fixes these patterns
A CAPA-forward platform fixes all three by design. The closure step is gated on a separate verification record. Role-based verifier setup excludes the action owner from verifying their own work. The CAPA is linked to its trigger record (audit finding, complaint, supplier issue, incident), and the trigger record carries the open/closed status of every CAPA against it.
The Verified Closure standard — preventive action evidence requirement
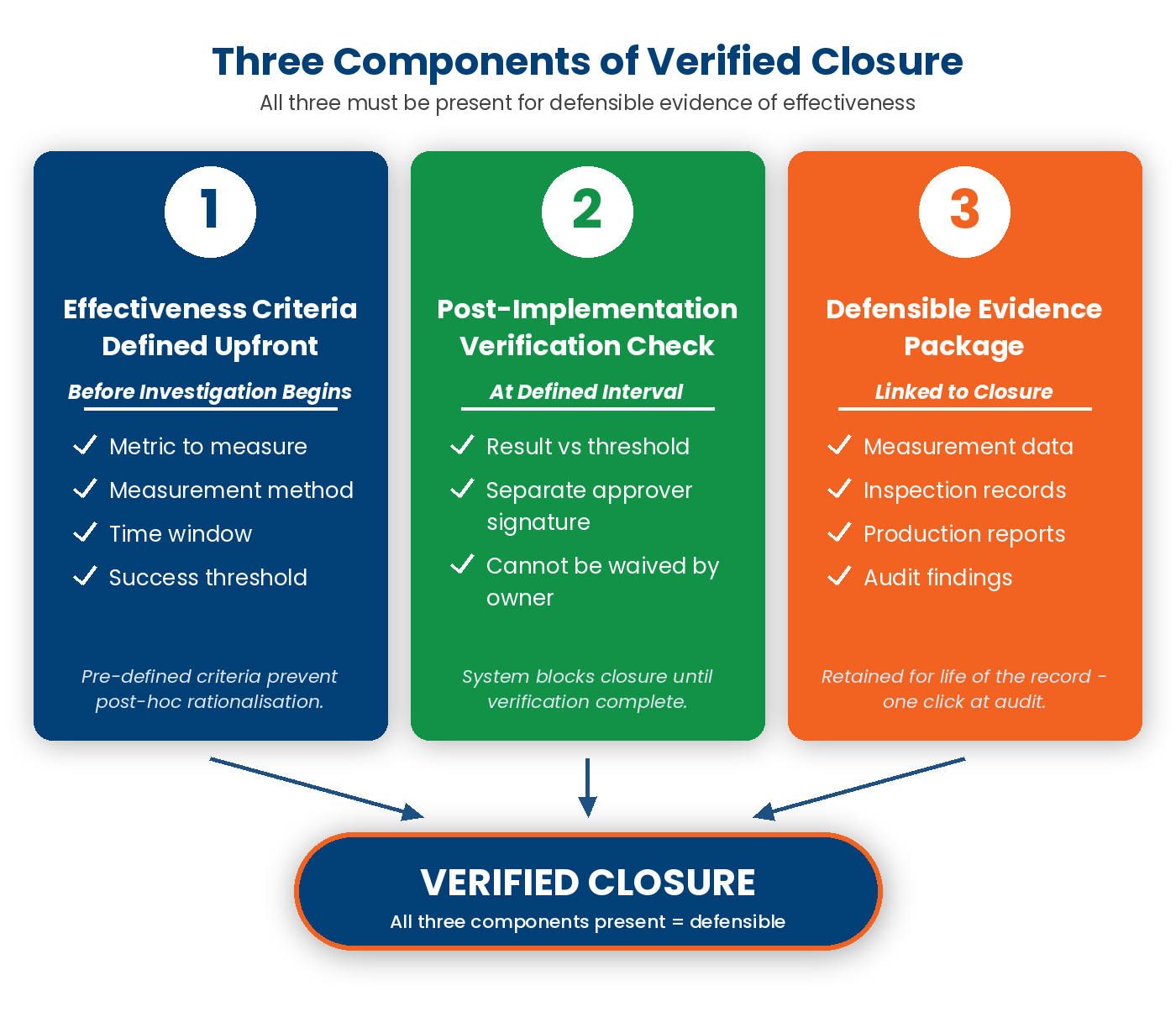
Verified Closure has three components, each with its own evidence requirement.
- Action Complete (with evidence). The corrective or preventive action has been performed and the owner has provided evidence of completion. That evidence is typically a photo, a signed work order, an updated procedure document, a re-trained operator list, or a parameter change recorded in a control plan. “Complete” without evidence is a claim, not a fact.
- Effectiveness Verified (by an independent reviewer). A second person — not the action owner — has reviewed the action and the evidence and validated that the action addresses the documented root cause. This is the gate most platforms skip. It is what ISO 9001 Clause 10.2.1.d (“review the effectiveness of any corrective action taken”) directly requires.
- Recurrence Prevented (over a defined window). The same control point, process, or product is sampled or audited inside a defined window (30, 60, 90, 180 days depending on action type and risk) to confirm the deviation has not recurred. This is where the discipline pays off. A CAPA closed in week 4 but still showing no recurrence in month 6 is a real closure. A CAPA closed in week 4 with recurrence in month 5 is a re-opened CAPA — and a finding under any competent surveillance audit.
A platform that enforces all three is what we mean by CAPA-forward. Two out of three is not enough; the recurrence-prevention check is the difference between a system that documents fixes and one that proves them.
Linking root cause to recurrence prevention
Root cause analysis is the bridge. A verified-closure workflow requires that the preventive action addresses the documented root cause, not just the symptom. If the root cause is “operator deviated from work instruction” and the preventive action is “remind operator of work instruction,” the verification step should fail. The action does not address the systemic cause (work instruction unclear, training inadequate, supervisor sign-off absent). Recurrence is predictable.
The pattern we look for is straightforward. Every CAPA has a documented root-cause method (5-Why, Fishbone, 8D). Every preventive action links directly to a root-cause line. And the verifier must confirm that link before closure. When the platform enforces this, recurrence rates drop below 10% — the mature-program benchmark — within two to three quarters. When it does not, recurrence rates sit at 30–50% and the team blames “the same five suppliers” or “the same two lines.”
Defensible evidence in CAPA
The output of a verified-closure workflow is defensible evidence — the same audit-trail standard we apply across the Certainty platform. Every CAPA record carries the timestamp of every action, the user identity, and the role of the action owner and the verifier (kept separate). It also carries the controlled-document revision against which the original deviation was raised, the photo or document evidence of the action, the verification record, and the recurrence-window check. Records are tamper-evident; changes are logged, not overwritten; deleted records persist in the audit trail.
An FDA inspector, an IATF Notified Body, an AS9100 surveillance auditor, or a Tier 1 customer auditor expects to see this when they ask “show me a recent CAPA from start to Verified Closure.” A CAPA program that cannot produce this trail — independent of the platform marketing it sits on — will fail a real audit, even if it passes the internal one. For a deeper treatment of defensible evidence as a cross-platform discipline, see our EHS software pillar guide.

Core CAPA capabilities
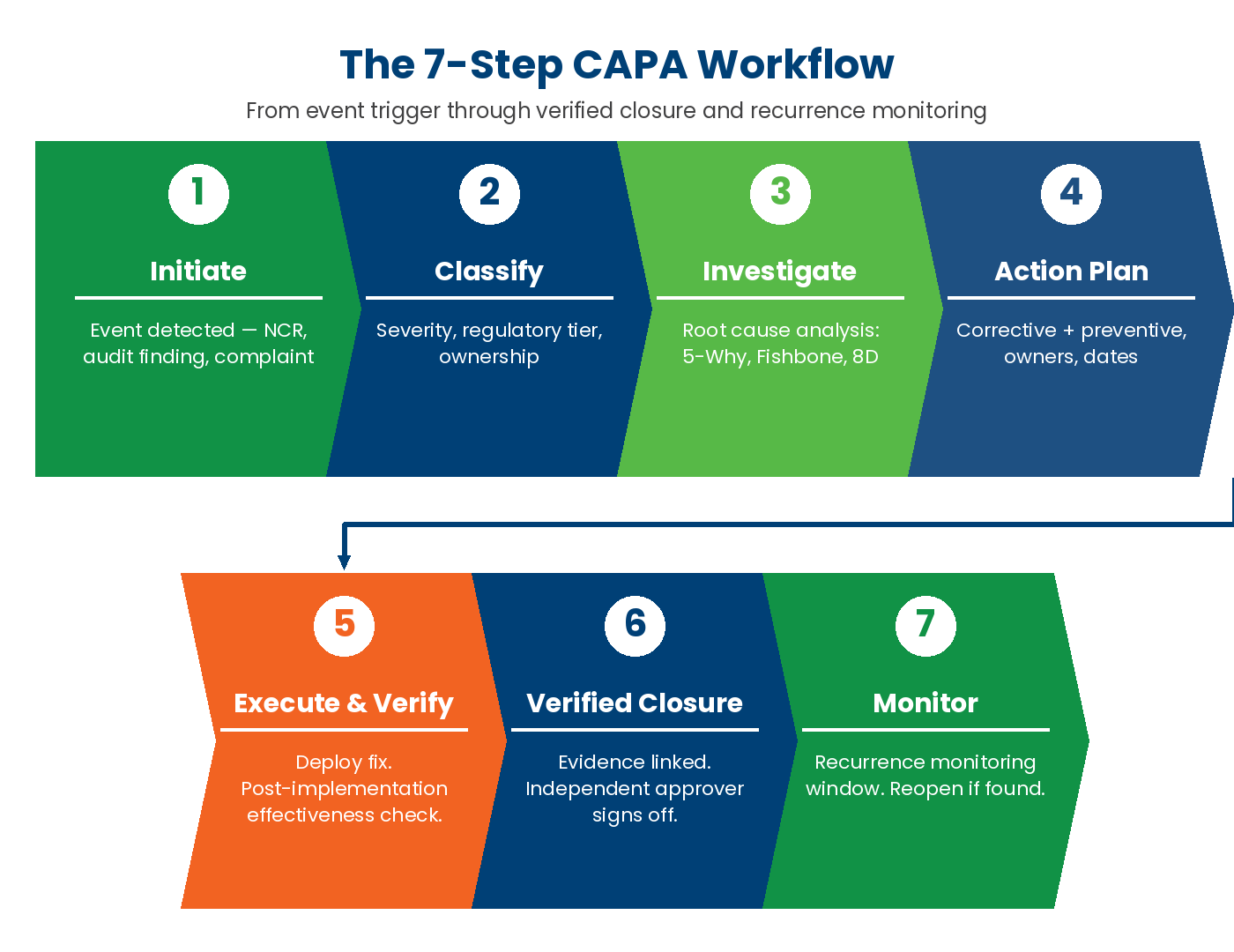
The minimum capability set for a real CAPA platform has converged. A platform that does not cover the six areas below is a form builder with a CAPA label, not a CAPA system. Those areas: multi-source initiation, formal investigation, corrective and preventive action plan, Verified Closure, closure with recurrence monitoring, and role-based reporting.
Initiation — incident, audit finding, customer complaint, supplier issue
CAPA is unusual among quality modules because the trigger event almost never originates in the CAPA tool itself. A modern CAPA platform must accept initiation from multiple upstream sources. These include an audit finding raised in the audit module, a customer complaint logged in the CRM, a supplier non-conformance from incoming inspection, a safety incident from the EHS module, and an internal observation from a layered process audit. Each one carries the link back to the originating record. Severity classification at initiation determines workflow path (safety-critical CAPAs route to expedited review; minor administrative CAPAs use a lightweight path).
The test: when a CAPA closes, can the audit finding, complaint, or incident that triggered it automatically reflect the closure status? If the trigger record stays “open” while the CAPA closes, the system is missing the link.
Investigation — 5-Why, Fishbone, 8D
A CAPA without a formal root-cause method is a guess. The platform must support multiple methods because different problem types demand different tools. Use 5-Why for simple deviations and Fishbone (Ishikawa) for multi-cause process problems. Use 8D for customer-facing automotive defects, fault tree for safety incidents with multiple contributing factors, and Pareto for prioritization across many similar findings.
The configuration test is whether the method is enforced by the workflow. A CAPA workflow that allows “root cause: see attached PDF” without a structured method is documentation theatre. A workflow that requires the user to declare the method, capture each step (e.g., the five “why” questions for a 5-Why, the six bones for a Fishbone), and link the resulting cause to a preventive action is a real investigation.
Action plan — corrective + preventive actions
Every CAPA needs both a corrective action (the immediate fix that resolves the current deviation) and a preventive action (the systemic change that prevents recurrence). They are not the same thing, and they are not optional. ISO 9001:2015 Clause 10.2 requires correction (10.2.1.a) and corrective action (10.2.1.b–c) with effectiveness review (10.2.1.d). The 2015 revision removed the standalone preventive action clause; prevention now lives in risk-based thinking at Clause 6.1. FDA QMSR / ISO 13485 § 8.5.3 and IATF 16949 § 10.2.3 retain the explicit preventive action requirement — and that is why a CAPA platform must support both corrective and preventive action as distinct workflow events.
The platform should require a separate field, owner, and due date for each. It should allow multiple corrective and preventive actions per CAPA — many real CAPAs have a quick containment action, a longer-term corrective action, and a process-level preventive action. And it should track each action’s status independently so the CAPA cannot close while any sub-action is still open.
Verification — proof of effectiveness (the Verified Closure step)
This is the gate. The verification step is a separate workflow event with its own assigned role (not the action owner), its own evidence requirement, and its own deadline. The verifier reviews the action evidence, evaluates whether it addresses the documented root cause, and decides whether the action is effective. A “not effective” decision routes the CAPA back to the action-plan step for re-work — not to the closure step with a note.
The recurrence-window check is the second half of verification. After the action is verified effective, the platform schedules a follow-up observation (audit, inspection, sample check, data review) inside a defined window appropriate to the action type and risk. The recurrence check carries the same evidence requirements as the verification: timestamp, reviewer, evidence type, outcome. This is Verified Closure in operating form.
Closure + recurrence monitoring
Closure is not the end of the record. A closed CAPA stays linked to its control point, its root cause, and its trigger source for the retention period of the regulation it falls under. That is 3 years for ISO 9001 baseline, 7+ for FDA-regulated industries, and 10+ for aerospace and medical device. Recurrence monitoring against the same control point continues for the lifetime of the product or process. If the same control point fires three CAPAs in eighteen months, the platform should escalate — the root cause is upstream of every one of them, and a new, deeper CAPA is required.
Reporting — open / aging / closed CAPAs by site, owner, severity
A CAPA platform without a reporting layer is an inventory. The reports quality and operations leaders need run constantly: open CAPAs by site, by owner, by severity; CAPA aging (median days open by severity class); CAPA closure cycle time trend (improving or degrading?); recurrence rate by site, by process, by supplier; CAPAs raised per quarter, per source (audit, complaint, supplier, incident). Role-based dashboards: shop-floor operator sees their open CAPAs; plant manager sees plant aging; corporate sees multi-site recurrence trend.
Drill-down from any KPI back to the underlying CAPA — and from any CAPA back to its trigger, root cause, and verification record — is not a nice-to-have. It is what a customer auditor or a regulator will ask for. If the platform requires a spreadsheet export to answer the question, the platform has failed.
CAPA in regulatory context
CAPA is named directly in every major quality and safety management standard — ISO 9001 Clause 10.2, IATF 16949 Clause 10.2.3 (problem solving / 8D), AS9100 Clause 10.2, FDA 21 CFR Part 820.100, and ISO 45001 Clause 10.2. The clauses below are the ones that show up most often in surveillance audits and regulatory inspections.
FDA cGMP / FDA QMSR (21 CFR Part 820, as amended February 2026) — medical device + pharma
The FDA originally introduced CAPA as a discrete subsystem in 21 CFR Part 820.100, Subpart J of the Quality System Regulation (QSR) for medical devices. As of 2 February 2026, the QSR was replaced by the Quality Management System Regulation (QMSR), which retains Part 820 as the regulation but incorporates ISO 13485:2016 by reference; CAPA flows through ISO 13485 § 8.5.2 (corrective action) and § 8.5.3 (preventive action) via that incorporation.
Section 820.100 (now part of the QMSR) historically required, and the QMSR continues to require via ISO 13485 § 8.5.2 and § 8.5.3, that manufacturers establish procedures for: analyzing data sources for non-conforming product and quality problems, investigating causes, identifying actions to correct and prevent recurrence, verifying or validating the action’s effectiveness, implementing changes, and disseminating information. CAPA is the first system FDA inspectors review during a medical-device manufacturing inspection. The QSIT four-subsystem inspection approach was discontinued on 2 February 2026. FDA updated its existing Compliance Program 7382.850 to align with the QMSR framework; the inspection programme continues under this revised compliance program. The inspection priority on CAPA is unchanged. For many investigators, the state of the CAPA program is a leading indicator of the state of the entire QMS.
For pharmaceutical manufacturers, the same CAPA discipline applies under cGMP regulations and is integrated with deviation management, change control, and complaint handling. Validation (IQ/OQ/PQ) of the CAPA platform is required for FDA-regulated environments, and electronic signatures must comply with 21 CFR Part 11.
ISO 9001:2015 — Clauses 10.1, 10.2, 10.3
ISO 9001:2015 Clause 10 (“Improvement”) contains the CAPA core requirement. Three sub-clauses matter:
| ISO 9001:2015 Clause | What it requires | CAPA module relevance |
|---|---|---|
| 10.1 — General | The organization shall determine and select opportunities for improvement | Trend analysis from closed CAPAs feeds opportunity register |
| 10.2 — Nonconformity and corrective action | React to nonconformity, evaluate need for action to eliminate causes, implement, review effectiveness, update risks | The core CAPA workflow — all six sub-clauses map to CAPA module functions |
| 10.3 — Continual improvement | The organization shall continually improve the suitability, adequacy, and effectiveness of the QMS | Recurrence-rate trend, CAPA aging trend, closure cycle time trend |
The Clause 10.2.1.d language — “review the effectiveness of any corrective action taken” — is the explicit basis for Verified Closure. ISO 9001 is unambiguous: signature-based closure without effectiveness review is non-conformant. Note that while ISO 9001:2015 removed the standalone preventive action clause (Clause 8.5.3 of the 2008 edition), the intent — preventing nonconformities before they occur — is fully retained through risk-based thinking at Clause 6.1, and certification bodies continue to assess this discipline.
IATF 16949 — automotive 8D
IATF 16949:2016 layers automotive-specific requirements on top of ISO 9001. Clause 10.2.3 (“Problem solving”) directly names structured problem-solving method — the 8D (Eight Disciplines) approach is the de facto standard, with many OEMs (GM, Ford, Stellantis, VW Group) mandating 8D format and timing for customer-facing defects. Typical OEM customer-specific requirements specify 24-hour containment (D3), 7-day root cause and corrective action (D4–D6), and 30-day verified preventive action (D7) with effectiveness check (D8).
A CAPA platform used in automotive must support 8D as a first-class template — not as a PDF attached to a generic CAPA record. Each discipline (D1 through D8) is a workflow step with its own owner, deadline, and evidence requirement. The verified-closure logic maps cleanly to D7 (preventive action) and D8 (congratulate the team — but only after effectiveness is proven).
AS9100 — aerospace
AS9100 extends ISO 9001 Clause 10.2 with aerospace-specific verification depth. AS9100D Clause 10.2 retains the ISO baseline and adds aerospace-specific requirements around supplier nonconformity, escape investigation, and effectiveness verification at the production-line level. Clause 10.2.1.h specifically addresses flow-down of corrective action requirements to suppliers — the aerospace addition most often cited in audits. Document retention extends to 10+ years for flight-safety parts; the CAPA record must remain reproducible for the lifetime of the part in service.
For aerospace suppliers, the CAPA platform must also handle escapes — defects that reach the customer — with explicit timeline expectations from prime contractors. Escape CAPAs typically run on an accelerated 8D-like workflow with customer-facing reports and direct effectiveness verification by the OEM’s supplier quality engineer.
ISO 45001 / OSHA — safety incident CAPA
Quality is not the only ICP that uses CAPA. ISO 45001 Clause 10.2 mirrors ISO 9001’s corrective-action requirement for occupational health and safety. Safety incidents — first-aid events, recordable injuries, near-misses, environmental releases — route into the same CAPA workflow with the same verified-closure discipline. The action owner is typically the EHS lead or the line supervisor. Verification falls to an independent EHS auditor or safety committee member. The recurrence window is often shorter — immediate re-inspection, 30-day re-observation — because safety-critical recurrence has higher consequence.
OSHA recordkeeping (29 CFR 1904) feeds directly into the same CAPA system in well-run organizations. Recordable injuries trigger CAPA initiation automatically. The same audit trail that satisfies an ISO 9001 surveillance audit satisfies an OSHA inspection — defensible evidence is the cross-platform standard.
Method integrations — 8D, 5-Why, Fishbone, Pareto

Root cause method is not religion. The right method depends on the problem class, the time available, and the audience for the resulting CAPA. A CAPA platform that supports all four below — 8D, 5-Why, Fishbone, and Pareto — and lets the team pick the right tool outperforms one that hard-codes a single methodology.
8D problem solving — when to use
8D (Eight Disciplines) originated at Ford in the 1980s and became the automotive industry’s de facto problem-solving framework. It is the right tool when the problem is customer-facing, the OEM is asking for a structured response, and the timeline is compressed. The eight disciplines run in sequence:
- D1 — Team. Form a cross-functional team with the right expertise.
- D2 — Problem. Describe the problem with data — what, where, when, magnitude.
- D3 — Containment. Stop the bleed. Quarantine product, hold suspect lots, alert downstream.
- D4 — Root cause. Investigate why the problem occurred using 5-Why or Fishbone.
- D5 — Permanent corrective action. Identify the action that eliminates the root cause.
- D6 — Implement. Execute the corrective action.
- D7 — Prevent recurrence. Institutionalize the fix by updating procedures, controls, FMEA, and control plans.
- D8 — Recognise. Formally acknowledge the team’s contribution, document and standardise the solution across similar products and processes, and capture lessons learned — but only after D7 effectiveness is verified.
For a deeper treatment of 8D in practice, see our standalone guide on 8D problem solving. A CAPA platform that supports 8D must enforce the sequence — you cannot close D5 before D4 is documented; you cannot close D8 before D7 is verified.
5-Why root cause analysis
5-Why is the right tool when the problem is straightforward and the root cause is one or two layers below the surface. A line operator misses a torque-spec — why? — operator was rushed — why? — supervisor reassigned them mid-shift — why? — staffing model didn’t account for the work-instruction’s setup time — why? — the standard time was last revised three years ago. Five questions, one root cause, one preventive action (update standard time and re-balance the line).
The discipline of 5-Why is that you must stop asking “why” when the answer is human nature or a generic abstraction (“operator error,” “lack of attention”). Instead, ask “why was the system designed to allow this?” A CAPA platform should structure the 5-Why as five linked fields with a final “root cause” field that captures the systemic answer. For a deep-dive walk-through, see our root cause analysis guide.
Fishbone / Ishikawa diagrams
Fishbone (also called Ishikawa, after Kaoru Ishikawa who formalized the technique) is the right tool when the problem has multiple potential causes that need to be enumerated before investigation. The classic six “bones” — Manpower, Method, Machine, Material, Measurement, Environment (the 6Ms) — give the team a structured way to brainstorm contributing factors before testing them.
Fishbone works best in a cross-functional CAPA team setting where the problem is process-level (yield drop, defect rate increase) rather than single-event. The CAPA platform should support the structure with a diagram-style input. Use six categories, multiple causes per category, and the ability to mark which causes were tested and which were ruled out by data.
Pareto analysis for CAPA prioritization
Pareto is the right tool when the question shifts. Instead of “what caused this CAPA,” ask “of the hundred CAPAs we closed last quarter, which root causes account for 80% of the volume?” A CAPA reporting layer should support Pareto by root-cause category: operator error, equipment malfunction, design flaw, supplier defect, material variation, and training gap. That view lets quality and operations leaders see where to spend the next CAPA investment. It also exposes systemic problems that single-CAPA investigation misses (twenty CAPAs across five product lines all rooted in the same control-plan parameter).
Pareto is the method that turns CAPA from a reactive tool into a continuous-improvement engine. A platform without it leaves the CI program guessing.
How to choose CAPA software — buyer’s framework
CAPA software selection failures usually come from one of five mistakes: buying a rigid vendor-coded workflow; ignoring multi-site rollup and escalation; under-specifying upstream integration with QMS and EHS; treating mobile and electronic signature as afterthoughts; or accepting an audit trail that won’t survive a 21 CFR Part 11 inspection. The framework below is built to prevent each of them.
Configurable workflow vs rigid template
The single biggest CAPA selection mistake is buying a platform with a rigid, vendor-coded CAPA workflow. Every organization has a slightly different CAPA process — different severity classes, different escalation rules, different method emphasis (8D-heavy in automotive, 5-Why-heavy in industrial, Fishbone-heavy in food). A platform that requires a vendor consultant to add a severity class, change an escalation rule, or modify the verification gate will fight your CAPA program for its entire life.
A flexible CAPA platform lets the quality team itself adjust the workflow — add or remove severity classes, change action-plan field requirements, modify the verification role, set the recurrence-window window per severity. The test in the demo: ask the vendor to add a new severity class, change an escalation timeline, and add a custom field to the investigation step in front of you. If they cannot, the platform is custom-coded under the hood.
Multi-site rollup + escalation rules
If you operate more than one site, CAPA must roll up across them. That means site-scoped CAPA visibility: Site A’s quality team sees Site A’s open CAPAs by default. Corporate roles get cross-site visibility — the QA Director sees aging across every plant. And recurrence detection runs across sites — the same root cause at two plants triggers a corporate-level CAPA, not two independent local ones.
Escalation rules are the multi-site corollary. A CAPA that misses its first deadline should escalate to the assigned owner’s manager. A CAPA that misses its verification gate should escalate to the QA Director. Any safety-critical CAPA past 48 hours should escalate to the VP Operations. These rules need to be configurable, role-based, and visible — so everyone knows when escalation is coming.
Integration with QMS, EHS, ERP
CAPA does not live alone. The platform must integrate with the upstream quality and safety modules — audit findings, customer complaints, supplier issues, safety incidents. It must also integrate with downstream business systems: ERP for parts and supplier master data, MES for process parameters, and BI for executive dashboards. Native integrations into the broader quality management software ecosystem matter more than they look on a feature list. A CAPA tool disconnected from the audit module is the source of the “CAPA in another database” failure pattern we described in Verified Closure.
The integration test is simple. When an audit finding is raised, can the platform automatically create a CAPA with the finding as its trigger record, the auditor as the initial assignee, and the controlled-document revision pre-populated? If yes, the integration is real. If no, the CAPA workflow is bolted on.
Mobile capture + signature support
CAPA evidence is captured where the work happens — the shop floor, the warehouse, the supplier site, the field service location. The platform must run on a tablet or phone with photo evidence at every step — containment photo at D3, action photo at D6, verification photo at D7. Electronic signature is required at the action-complete and verification gates. Offline capture (welding bay, refrigerated room, supplier site without Wi-Fi) with sync-on-reconnect is the modern minimum.
A desk-only CAPA platform produces a desk-only CAPA program — actions documented from memory, evidence reconstructed at week’s end, photos that someone meant to take.
Audit trail + electronic signature compliance (21 CFR Part 11)
The CAPA audit trail must be defensible under regulatory scrutiny — FDA inspection, IATF surveillance, AS9100 surveillance, customer audit. That means every record carries timestamp, user identity, action, controlled-document revision, and (where required) Part 11-compliant electronic signature with binding to the underlying record. Records are tamper-evident; changes are logged; deleted records persist with the deletion event recorded.
For FDA-regulated industries (medical device, pharma, biotech), 21 CFR Part 11 compliance is not optional. The platform must support validated electronic signatures, role-based access controls with documented procedures, and audit-trail export in human-readable form. Validation (IQ/OQ/PQ) packages must be available from the vendor for regulated deployments.
See how Certainty supports Verified Closure
Configurable · Mobile-first · Multi-site · Verified Closure on every CAPA · One audit trail, every standard
See Certainty’s audit management softwareCAPA rollout roadmap (6 steps)
A focused CAPA rollout does not need to be a multi-quarter project. The six-step roadmap below is what we see succeed across quality and EHS teams, completed in 6 to 12 weeks for a single-site rollout and 3–4 months for a multi-site enterprise.
Step 1 — Map current CAPA process and find the gaps
Before any platform is configured, document how CAPA actually runs today. Where do CAPAs originate (audit, complaint, supplier, incident, internal observation)? Who classifies severity? Who assigns owners? What method is required for which class? How is verification handled — and is it independent? What is the recurrence-window check, if any? What is today’s median cycle time, aging profile, and recurrence rate?
Mark each step as mature, broken, or absent. The output is a one-page current-state map. The gaps you find here — most commonly, missing verification step, no recurrence check, fragmented trigger sources — are the rollout backlog.
Step 2 — Define severity + classification matrix
Severity drives the workflow. Define classes that map to your real operational risk — for example: Safety-critical (immediate), Customer-facing major, Customer-facing minor, Internal major, Internal minor, Administrative. For each class, set five things: time to containment, time to root-cause complete, and time to Verified Closure. Specify the required method (8D mandatory for customer-facing, 5-Why acceptable for internal minor). And specify the recurrence-window check (90 days for customer-facing, 180 for internal major, none for administrative).
Get sign-off from QA, EHS, Operations, and Compliance before configuring the platform. Severity is the most-argued part of any CAPA program; agreeing it upfront prevents months of re-work.
Step 3 — Configure investigation methods
Stand up the method templates the business actually uses. For automotive, 8D is non-negotiable; configure it as a first-class workflow with D1–D8 as sequential gates. For general manufacturing, 5-Why and Fishbone with a methodology-selector at investigation start. In food and pharma, add fault-tree analysis for safety-critical or recall-risk events. Then link each methodology’s root cause to the preventive-action step so the platform can later verify the preventive action addresses the cause.
This is the step where most quality teams over-build. Keep templates short and observable; you can add complexity in a v2.
Step 4 — Stand up Verified Closure rules
Configure the verification gate as a separate workflow step with its own role, evidence requirement, and deadline. Define the verifier role to exclude the action owner and their direct reports. Specify what evidence is required for each action type — photo for physical change, signed procedure for document change, training record for competency change, sample data for parameter change. Configure the recurrence-window check as a scheduled follow-up task with its own assignee (typically the original auditor or a peer reviewer) and its own evidence type.
Run the configuration through a tabletop exercise with three real, recently-closed CAPAs from the legacy process. If the platform would re-open any of them under the new rules, you have built the system correctly.
Step 5 — Pilot with 1 site / 1 product line
Roll out to one site and one product line for 30 days before broader deployment. Run every CAPA — quality, safety, supplier — through the new platform. Hold the legacy process in parallel for the first 14 days; expect 2× workload temporarily. After 30 days, review with the site team: which workflow steps slowed people down, which evidence requirements were unrealistic, which severity classes need adjustment. Iterate the configuration before scaling.
The pilot is also where you learn whether the verification role is staffed. Verified Closure is impossible if there is no one with the time, authority, and independence to verify. If the pilot reveals this gap, fix the staffing before you scale — adding a platform on top of an unstaffed verifier role just documents the gap.
Step 6 — Scale + measure (open / aging / recurrence rate)
Once the pilot is stable, scale to the remaining sites in 30-day waves. At each wave, monitor three metrics: open CAPA count (is the backlog growing or stable?), CAPA aging (median days open by severity — improving or degrading?), and recurrence rate (% of CAPAs closed in the last 12 months that have recurred at the same control point). The mature-program benchmark is recurrence rate below 10%; internal programme assessments and quality consulting benchmarks commonly report recurrence rates of 30–50% in organisations without formal effectiveness verification steps, though published industry data on this specific metric is limited — organisations should establish their own baseline as a first step.
Review monthly with the operations leadership team, quarterly with the executive team, and annually as part of formal Management Review per ISO 9001 Clause 9.3. CAPA performance is one of the most direct leading indicators of QMS health — if it improves, COPQ follows two to three quarters later.
Key Takeaways
- CAPA software is the digital system of record for the closed-loop workflow that takes a non-conformance — from any source — through investigation, root-cause analysis, corrective and preventive action, and Verified Closure with proof of effectiveness.
- Verified Closure is Certainty-owned vocabulary and the discipline that separates a CAPA program that learns from one that repeats findings. It treats “action complete” and “action effective” as two distinct events, with independent verification and a recurrence-window check.
- The six core capabilities of a real CAPA platform: multi-source initiation, structured-methodology investigation, corrective + preventive action plan, Verified Closure, recurrence monitoring, and role-based reporting with drill-down.
- CAPA is named directly in every major quality and safety standard: ISO 9001 Clause 10.2, IATF 16949 Clause 10.2.3 (problem solving / 8D), AS9100 Clause 10.2, FDA 21 CFR Part 820.100, and ISO 45001 Clause 10.2.
- Method selection matters: 8D for customer-facing automotive defects, 5-Why for internal deviations with shallow root causes, Fishbone for multi-cause process problems, Pareto for prioritization across many CAPAs.
- The buyer’s framework: flexible workflow (not vendor-coded templates), multi-site rollup with escalation, integration with QMS and EHS upstream, mobile capture with offline support, 21 CFR Part 11-grade audit trail.
- A focused CAPA rollout runs 6–12 weeks for a single site, 3–4 months multi-site — not the multi-quarter projects validated eQMS vendors still quote.
- The CAPA metric that matters most is recurrence rate: % of CAPAs closed in the last 12 months that have recurred at the same control point. Mature-program benchmark is below 10%; internal programme assessments and quality consulting benchmarks commonly report recurrence rates of 30–50% in organisations without formal effectiveness verification steps, though published industry data on this specific metric is limited — organisations should establish their own baseline as a first step.
Frequently Asked Questions (FAQs)
What does CAPA stand for?
CAPA stands for Corrective and Preventive Action. It is the closed-loop workflow that takes a documented non-conformance through investigation, root-cause analysis, corrective action (fixing the immediate issue), preventive action (preventing recurrence), and Verified Closure with proof of effectiveness. The term originated in the FDA’s Quality System Regulation (21 CFR Part 820.100) for medical devices. As of 2 February 2026, the QSR has been replaced by the Quality Management System Regulation (QMSR), which retains the Part 820 location but incorporates ISO 13485:2016 by reference — CAPA requirements now flow through ISO 13485 § 8.5.2 (corrective action) and § 8.5.3 (preventive action). The CAPA concept is also embedded in ISO 9001 (Clause 10.2 — correction and corrective action; ISO 9001:2015 removed the standalone preventive action requirement and embedded prevention in risk-based thinking at Clause 6.1), IATF 16949 (Clause 10.2.3 — 8D problem solving), AS9100 (Clause 10.2), and ISO 45001 (Clause 10.2).
What’s the difference between corrective and preventive action?
A corrective action fixes the immediate, observed deviation — the part is out of spec, so re-work the part; the procedure was not followed, so re-train the operator. A preventive action addresses the systemic cause so the same deviation cannot recur — the spec was unclear, so revise the work instruction; the training program omitted the step, so update the training matrix and re-train all affected operators. ISO 9001:2015 Clause 10.2 covers correction (10.2.1.a) and corrective action (10.2.1.b–c) with effectiveness review (10.2.1.d). The 2015 revision of ISO 9001 removed the standalone preventive action clause that existed in 2008 — prevention now lives in risk-based thinking at Clause 6.1. The explicit preventive action requirement is retained under FDA QMSR / ISO 13485 § 8.5.3 and IATF 16949 § 10.2.3. Corrective and preventive action are different events with different owners, different evidence, and different deadlines.
Is CAPA software required for ISO 9001 or FDA compliance?
No — neither ISO 9001:2015 nor the FDA QMSR (21 CFR Part 820 as amended February 2026) directly require software. Both standards require a documented CAPA process with formal investigation, root-cause analysis, action, and effectiveness review. These can be operated on paper or spreadsheet. In practice, the audit-trail, electronic-signature, and reproducibility requirements of a defensible CAPA system are difficult to satisfy without software at any meaningful scale. For multi-site or FDA-regulated environments, satisfying them efficiently is impossible. Most CAPA programs that fail a regulatory or customer audit fail not because the process is wrong but because the evidence cannot be reproduced under scrutiny.
How long should a CAPA take to close?
CAPA cycle time depends on severity. Safety-critical CAPAs (immediate hazard, customer escape) should reach containment within 24 hours and Verified Closure within 30 days. Customer-facing major CAPAs (defect reaching the customer, OEM-reported issue) typically run 24-hour containment, 7-day root cause, 30-day Verified Closure on an 8D framework. Internal major CAPAs run 30–60 days median. Internal minor and administrative CAPAs run 30–90 days. The mature-program benchmark for median cycle time is below 30 days for routine quality CAPAs and below 10 days for safety-critical. A median CAPA aging above 90 days is a leading indicator of a stalled program.
What is “Verified Closure” and why does it matter?
Verified Closure is the discipline of treating “action complete” and “action effective” as two separate events in a CAPA workflow. An action is complete when the assigned owner has performed it and provided evidence. A CAPA is closed only after an independent verifier confirms the action addresses the documented root cause, and a recurrence-window check (typically 30–180 days depending on action type) confirms the deviation has not recurred. Verified Closure is what ISO 9001 Clause 10.2.1.d directly requires when it asks organizations to “review the effectiveness of any corrective action taken.” It is Certainty Software’s defined standard, and it is the test that separates a CAPA program that learns from one that repeats the same findings year after year.
What’s the difference between CAPA and NCR (non-conformance)?
An NCR (Non-Conformance Report) is the documented deviation itself — a single event: a part out of spec, a procedure not followed, an audit finding raised. A CAPA is the closed-loop workflow that takes the NCR through investigation, root-cause analysis, corrective and preventive action, and Verified Closure. The NCR is the trigger; the CAPA is the system that resolves it. Many platforms collapse the two into one record, which is acceptable for low-severity events but problematic for major non-conformances where multiple corrective actions, multiple methods, and multi-stage verification are required. A well-designed CAPA platform keeps the NCR as the trigger record and the CAPA as the workflow shell, linked but distinct.
When should I use 8D vs 5-Why?
Use 8D when the problem is customer-facing, the OEM or regulator expects a structured response, the timeline is compressed, and a cross-functional team is required. 8D is the de facto standard in automotive (mandated by IATF 16949 Clause 10.2.3 and most OEM customer-specific requirements) and is increasingly common in aerospace and medical device. Use 5-Why when the problem is internal, the root cause is one to three layers below the surface, and a single investigator with line-supervisor input can resolve it. 5-Why is the right tool for the majority of internal quality CAPAs and most ISO 45001 safety incidents. Use Fishbone (Ishikawa) when multiple potential causes need to be enumerated before testing — typically process-level problems with several contributing factors.
How does CAPA software integrate with audit management?
A well-integrated CAPA platform receives audit findings directly from the audit module as CAPA initiation events — no manual re-entry, no copy-paste, no broken link. The audit finding stays linked to the resulting CAPA for the lifetime of the record. When the CAPA closes, the finding’s status updates automatically. And when the same control point fails again in a future audit, the system flags the prior CAPA on that control point as part of the new finding’s context. This is the difference between a CAPA module and a CAPA database. The platform must also feed verified-closure data back to the audit module for trend reporting — repeat findings at the same control point are the most-watched KPI in any audit management software program.
What is CAPA-forward CAPA software?
CAPA-forward is Certainty Software’s category description for a platform that treats corrective and preventive action as the first-class workflow rather than a feature inside a larger module. In a CAPA-forward platform, every audit finding, customer complaint, supplier issue, and safety incident routes directly into a CAPA workflow with required investigation methodology, action plan, and Verified Closure gate. The architecture inverts the older pattern in which CAPA was a back-office form attached to other modules — making CAPA the spine of the quality and safety system, not a side effect of it.
What does CAPA software typically cost?
Standalone CAPA platforms range from US$8,000 to US$60,000 annually for single-site mid-market deployments, depending on user count and module depth. CAPA capability embedded inside a broader quality or safety management platform – as is the case with Certainty Software – is usually included in the platform fee at no extra cost. This is one reason the platform-with-CAPA-module pattern has overtaken the standalone-CAPA-tool pattern for most buyers.
Validated CAPA systems for FDA-regulated industries must satisfy 21 CFR Part 11 (electronic records and signatures) and undergo computer system validation. The traditional IQ/OQ/PQ approach is increasingly supplemented or replaced by a risk-based Computer Software Assurance (CSA) approach per FDA’s 2025 final Computer Software Assurance guidance (issued 24 September 2025 for devices; pharmaceutical final guidance February 2026), which retains the risk-based, fit-for-intended-use approach first proposed in the 2022 draft. Total cost runs $50,000–$250,000 annually with rollout and validation adding a similar one-time amount. The relevant comparison is total cost of ownership including the cost of not having Verified Closure — repeat findings, customer escapes, regulatory observations.
How does CAPA software support 21 CFR Part 11?
For FDA-regulated industries, the CAPA platform must support 21 CFR Part 11 electronic-record and electronic-signature requirements. That means five concrete things. First, unique user authentication with role-based access. Second, an audit trail capturing every record creation, modification, and deletion with timestamp, user, and action. Third, electronic signatures bound to the underlying record — not just a typed name. Fourth, record retention and reproducibility for the regulatory window (typically 2 years past device or product life). Fifth, validation packages (IQ/OQ/PQ) showing the system performs as specified. A CAPA platform without these features cannot be used in an FDA-regulated environment, regardless of how good the workflow is.
How is CAPA software different from a generic ticketing system?
A ticketing system (Jira, Zendesk, ServiceNow) tracks work items with assignees, deadlines, and status fields. A CAPA platform does that and: enforces formal root-cause method (5-Why, Fishbone, 8D); requires linked trigger sources (audit, complaint, supplier, incident); enforces a separate verification step with role-based exclusion (action owner cannot verify their own work); captures recurrence-window checks against the same control point; produces a defensible audit trail that satisfies ISO 9001, FDA, IATF, AS9100, and ISO 45001 surveillance; supports electronic signatures bound to records per 21 CFR Part 11. Using a generic ticketing system for CAPA produces ticketing-quality CAPA — closed on signature, no verified effectiveness, no recurrence prevention, no defensible audit trail.
Related resources
CAPA, root cause, and methodology
- CAPA Process: 6 Steps for Improved Quality Management
- Corrective Action Examples
- A Comprehensive Guide to Root Cause Analysis
- 8D Problem Solving: A Guide for Businesses
- Implementing DMAIC Process: A Guide
- 4 Ways You Can Prevent Non-Conformances
Sibling and parent pillars
- Quality Management Software: The Complete Guide
- EHS Software: The Complete Guide
- Layered Process Audits: The Complete Guide
- Supplier Risk Management: The Complete Guide
Standards and regulatory references
- IATF 16949: Automotive Quality Management Standard
- What is the ISO 9001 Audit?
- ISO 19011: A Comprehensive Guide to Quality Management Auditing
- Quality Management System Audit Checklist
Certainty platform resources
- Audit Management Software
- Quality Audits Solution
- Supplier Audits Solution
- Poka Yoke: The Ultimate Guide to Error Prevention
- Continuous Improvement Assessment
Ready to bring Verified Closure to your CAPA program?
Most CAPA programs do not fail because the team is unqualified or the standards are unclear. They fail because closure is signature-based, verification is administrative, and recurrence is invisible. A CAPA-forward platform with Verified Closure built into every workflow changes the trajectory in one quarter.
See how Certainty’s configurable, mobile-first platform runs CAPA across quality, EHS, and supplier programs on one platform — with Verified Closure built into every workflow.


