
Quality Management Software: The Complete Guide
Quality Management Software (QMS) is the digital system of record for how a manufacturer plans, controls, audits, and continuously improves product and process quality. A modern QMS — sometimes called an eQMS, or electronic quality management system — replaces paper checklists, scattered spreadsheets, and disconnected point tools with one defensible source of truth for audits, inspections, non-conformances, and corrective and preventive actions (CAPA) — the execution and CAPA core that connects to the document control, training, and ERP systems that complete the quality picture. It is the backbone that supports ISO 9001:2015, IATF 16949, AS9100, FDA cGMP, and customer-specific quality requirements, and the platform that connects shop-floor data capture to executive-level quality KPIs.
Today, the QMS market has fragmented into two camps. On one side, heavyweight eQMS platforms built for regulated pharma and medical device — long implementations, expensive consultants, rigid workflows. On the other, lightweight “form builder” tools that handle simple inspections but fall apart at scale, audit-readiness, and corrective-action closure. The configurable, mobile-first, CAPA-forward category sits in between — and that is the category most quality and operations leaders are buying today.
QMS at a glance
| What it is | A configurable platform that runs the quality processes a manufacturer is audited against — documents, audits, CAPA, training, supplier quality, risk |
| Who uses it | QA Managers, Plant Managers, QA Directors, VPs of Operations, multi-site quality leaders |
| Core capabilities | Document control, audit management, non-conformance + CAPA, training, risk-based planning, multi-site reporting |
| Standards covered | ISO 9001:2015, IATF 16949, AS9100, FDA cGMP / 21 CFR Part 11, HACCP / GFSI, ISO 13485 |
| Deployment | Cloud (SaaS) — typically multi-site, multi-language, mobile-first; on-premise is now rare outside heavily regulated pharma |
| Outcome | Lower Cost of Poor Quality (COPQ), higher first-pass yield (FPY), defensible audit evidence, faster customer audit prep |
Table of contents
- What is quality management software?
- Core capabilities of a modern QMS
- CAPA-forward QMS — what it means
- Standards covered by QMS software
- How to choose QMS software — the buyer’s framework
- QMS implementation roadmap (6 steps)
- Defensible evidence and audit readiness
- QMS KPIs and benchmarks
- QMS across industries
- Frequently Asked Questions (FAQs)
- Related resources

What is Quality Management Software?
Quality management software (QMS software) is the digital system of record a manufacturer uses to plan, control, audit, and continuously improve product and process quality across one site or many. It manages audits and inspections, captures non-conformances, drives CAPA to Verified Closure, and reports performance against the standards the business is certified to — working alongside the document-control and training systems that round out the quality system. In short, QMS software is what makes a Quality Management System run — without it, the “system” is a collection of binders, spreadsheets, and tribal knowledge.
The acronym “QMS” has two everyday meanings. Internal quality teams use “QMS” to describe the management system itself — the policies, procedures, and processes that govern quality (per ISO 9001:2015). Software buyers use “QMS” or “qms software” to describe the platform that operationalizes that system. This guide uses the software meaning unless otherwise noted.
QMS vs eQMS vs Quality Audit Software — disambiguation
Three terms collide in this category. They are related but not interchangeable.
- QMS software — the umbrella category. Any digital platform that manages multiple quality processes (documents, audits, CAPA, training, supplier quality) under one roof. Most buyers searching “quality management software” are looking for this.
- eQMS (electronic Quality Management System) — historically a label for heavyweight, validated platforms built for pharma, medical device, and other FDA-regulated industries. eQMS implementations are usually 6–18 months, run by IT and Validation teams, and priced in six figures. The category is now broadening — many “QMS software” platforms call themselves eQMS interchangeably.
- Quality audit software — a narrower category focused on conducting audits and inspections (internal audits, supplier audits, layered process audits, food safety audits). Audit software is one capability inside a full QMS; standalone audit tools are common as a first step on the path to a complete QMS.
In practice, the simplest way to choose between them: if you need to run audits today and CAPA next quarter, start with audit software that has a credible upgrade path. If you need to replace document control, training, audit, and CAPA all at once because of a regulatory deadline, you are buying a full QMS. If you are validated pharma or medical device with rigid 21 CFR Part 11 requirements, eQMS is the right vocabulary.
Who uses QMS software
Today, QMS software is bought and used by quality and operations leaders, but the buying committee is broader than that.
- QA Manager / Quality Manager. Day-to-day owner of the system. Configures audits, manages document control, runs CAPA, prepares for customer and certification audits.
- Plant Manager / Operations Manager. Uses the platform to see real-time non-conformance, escape, and audit-completion data across their floor. Sponsors Layer 3 and Layer 4 audit participation.
- QA Director / VP Quality. Multi-site rollup, KPI reporting to the executive team, certification posture across the enterprise.
- VP of Operations / COO. Funds the platform. Cares about Cost of Poor Quality, customer escapes, and audit-readiness across all plants.
- IT / Information Security. Validates the platform’s authentication, audit trail, data residency, and integration footprint with ERP, MES, and SAP.
For multi-site organizations, the buying committee typically includes Quality, Operations, IT, and Finance — and the buying process usually takes 8–16 weeks for a configurable platform versus 6–12 months for a traditional eQMS.
Industries served
Additionally, QMS software is used across every industry where consistent process execution determines product quality, regulatory compliance, or customer satisfaction. The largest concentrations are in:
- Automotive and automotive supply — IATF 16949 and customer-specific OEM requirements
- Aerospace and defense — AS9100 and AS9120
- Food and beverage — HACCP, SQF, BRCGS, FSSC 22000 (GFSI-recognized standards)
- Medical device — ISO 13485 and the FDA QMSR (21 CFR Part 820 as amended February 2026, incorporating ISO 13485:2016 by reference)
- Pharmaceutical and biotech — cGMP, 21 CFR Part 11, ICH Q10
- Industrial manufacturing — ISO 9001 as a baseline with customer-specific add-ons
Notably, the same platform serves all of these industries when it is genuinely configurable. Industry-specific QMS platforms (medical-device-only, food-only) exist, but they are gradually losing share to multi-industry, configurable platforms because most manufacturers operate in more than one regulatory regime — a Tier 1 automotive supplier may also need AS9100 for aerospace work, and a food processor may also need ISO 14001 environmental.
Core capabilities of a modern QMS
QMS spans a spectrum — from focused audit-and-CAPA execution platforms to full eQMS suites that also own document control, a training LMS, and supplier-master data. Few manufacturers need all of it in one box, and many deliberately run a best-of-breed stack: a configurable audit-and-CAPA platform that integrates with their document-control, training, and ERP systems. The modules below make up the category; the question is which you run as one system and which you integrate. Certainty’s wedge is the audit-and-CAPA core — and a CAPA-forward architecture that most full suites bolt on as an afterthought.
Document control and version management
At its core, document control is the traditional foundation of a full eQMS — and where heavyweight programs most often stall. Many manufacturers run document control in a dedicated system and connect it to a configurable audit-and-CAPA platform, rather than buying one monolith for both. A capable document-control function handles controlled documents (procedures, work instructions, forms, control plans) with explicit version control, change history, electronic signatures, training-on-revision, and retention rules that satisfy ISO 9001:2015 Clause 7.5. The test that separates real document control from a shared drive: when a procedure is revised, the system automatically marks affected operators as “untrained on the current version” until they re-acknowledge — and audits cannot record compliance against an obsolete revision.
Audit management — LPA, layered, internal, and supplier
A modern QMS runs all audit types in one platform — internal audits against ISO 9001, layered process audits per CQI-8, supplier audits, food safety audits, customer audits — with a shared question library, shared CAPA workflow, and shared evidence repository. The advantage is not just consolidation; it is that a finding raised in a layered process audit can roll up against the same CAPA, the same root-cause analysis, and the same trend report as a finding raised by a Notified Body during certification. One audit trail, every standard.
Non-conformance and CAPA — with Verified Closure
This is the section most QMS deployments under-invest in, and the one with the highest leverage. A non-conformance must trigger a CAPA workflow with assigned owner, due date, root-cause analysis, corrective action, preventive action, and — critically — Verified Closure with effectiveness evidence. The verification step is what auditors look for and what most platforms skip. We treat this as a brand-defining capability and have made it a DefinedTerm in this guide’s schema (see CAPA-forward QMS below).
Training and competency tracking
Equally important, training is the connector between document control and audit. When a document is revised, affected roles must be retrained; when an operator is observed performing a task, the system must verify they are trained on the current revision. A modern QMS tracks training matrices by role, automatic re-training triggers on document revision, competency assessments tied to job descriptions, and expiration-driven retraining for time-bound qualifications (welding, forklift, internal auditor).
Risk-based quality planning
As a result, ISO 9001:2015, IATF 16949, and AS9100 all moved to a risk-based approach. The QMS must capture risk assessments, link risks to controls (FMEA, Control Plan, work instruction), and route preventive actions when risks change. For automotive, this includes Process FMEA, Control Plans, and PPAP package management. In aerospace, Key Characteristics and First Article Inspection serve the same purpose. Medical device manufacturers add design controls and risk management per ISO 14971.
Reporting and dashboards — multi-site rollup
In practice, quality leaders run on lagging indicators (COPQ, FPY, customer PPM, complaints) and leading indicators (audit-completion rate, CAPA closure cycle time, training completion). A modern QMS provides role-based dashboards: shop-floor operator sees today’s audits and open NCs; plant manager sees plant-wide non-conformance and CAPA aging; corporate sees multi-site rollup with site comparison. The platform must support drill-down from any KPI to the underlying audit, finding, or action — without exporting to spreadsheet.
Supplier quality and incoming inspection
For manufacturers buying parts, raw material, or sub-assemblies, supplier quality is part of the QMS. Capabilities include supplier qualification, periodic re-assessment, incoming inspection plans, supplier corrective-action requests (SCAR) with Verified Closure, and supplier scorecards. Multi-tier visibility (supplier of supplier) is becoming a buyer requirement under emerging due-diligence regulations like CSDDD.
CAPA-forward QMS — what it means
Term: A CAPA-forward QMS is a quality management platform in which corrective and preventive action is the first-class workflow, not a feature in a back-office menu. Every audit finding, non-conformance, customer complaint, and supplier issue routes directly into a CAPA that requires assigned owners, root-cause analysis, action evidence, and Verified Closure with effectiveness checks.
Most QMS deployments get the document-control and audit modules right and fail at corrective action. That is the single biggest reason ISO 9001 internal audits find the same non-conformances year after year. A CAPA-forward QMS inverts the architecture: the CAPA workflow is the core, and audits, NCRs, complaints, and supplier issues are inputs to it.
Why most QMS deployments fail at corrective action
In practice, three patterns repeat across organizations. First, CAPA is treated as a form — a description, a root cause field, an action — without enforced workflow, due dates, or escalation. Findings sit in “open” status indefinitely. Second, closure is signature-only — the assigned owner marks the action complete and the CAPA closes, with no verification step. Three months later the same finding recurs and the audit trail shows it was already “fixed.” Third, CAPA data is invisible to the rest of the QMS — the audit module doesn’t know that the auditee has three open CAPAs against the same control point, and the supplier scorecard doesn’t reflect the supplier’s open SCARs.
The Verified Closure standard
Verified Closure is the discipline of treating “complete” and “effective” as two different events. An action is complete when the owner has performed it and provided evidence. A CAPA is closed only after a second verification — typically a follow-up audit, a sample check, or a fresh data point demonstrating the action worked. Verified Closure is what auditors look for under ISO 9001 Clause 10.2 and IATF 16949 Clause 10.2.3 (problem solving). It is also what differentiates organizations that learn from their findings from those that repeat them.
Linking CAPA to root cause and recurrence prevention
In practice, a mature CAPA links every action to a documented root-cause analysis using a structured method — 5 Whys, fishbone diagram, 8D, or DMAIC for complex problems. The recurrence-prevention test is whether the action addresses the root cause or only the symptom. A QMS that supports this discipline forces the auditor or owner to declare the root cause method, document the cause, propose the action, and demonstrate via trend data that recurrence has stopped. For a deep dive on this workflow, see our CAPA software pillar guide.
Standards covered by QMS software
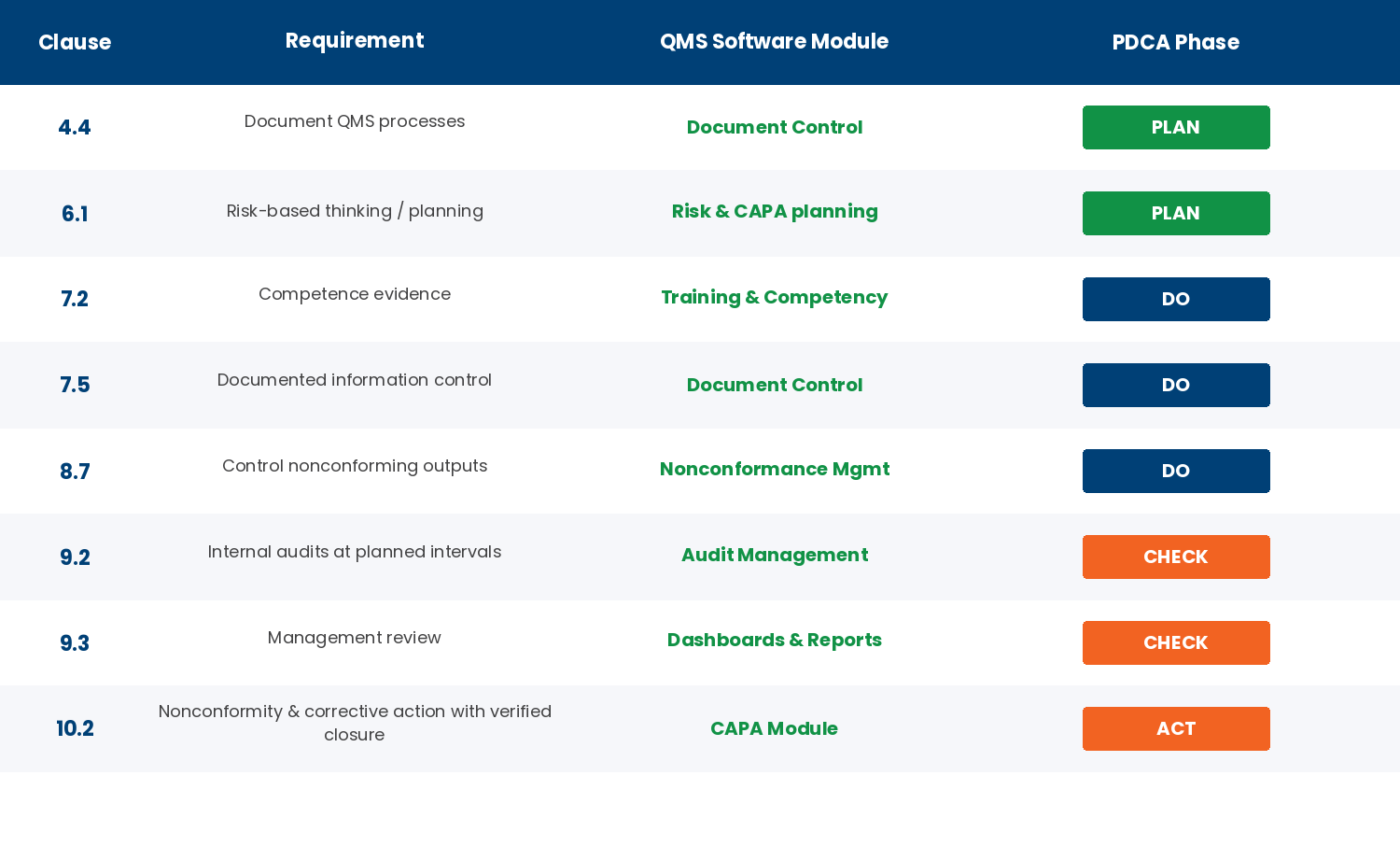
A modern QMS supports the major manufacturing and regulated-industry standards in one configurable platform. The standards below cover the majority of QMS use cases — ISO 9001:2015, IATF 16949, AS9100, FDA cGMP / 21 CFR Part 11, ISO 13485, and HACCP / GFSI.
ISO 9001:2015 — the umbrella standard
ISO 9001:2015 is the foundational quality management standard, applied across every manufacturing and service industry. Its ten clauses map directly to QMS modules:
| ISO 9001:2015 Clause | What it covers | QMS module |
|---|---|---|
| 4 — Context | Organizational context, interested parties, scope | Document control, policy management |
| 5 — Leadership | Quality policy, roles, customer focus | Document control, management review |
| 6 — Planning | Risk and opportunity, objectives, change | Risk register, KPI dashboards (note: ISO 9001:2015 moved preventive action from the standalone Clause 8.5.3 of the 2008 edition into risk-based thinking here at Clause 6.1; the intent — preventing nonconformities before they occur — is fully retained and assessed by certification bodies) |
| 7 — Support | Resources, competence, awareness, documents | Training, document control, equipment |
| 8 — Operation | Operational planning, requirements, design, supplier control | Audit, supplier quality, NCR |
| 9 — Performance | Monitoring, internal audit, management review | Internal audit, KPI dashboards |
| 10 — Improvement | Non-conformance, correction, corrective action, continual improvement | NCR, root-cause analysis |
In practice, a QMS purchase decision should map every must-have requirement to a clause and a module. If the platform cannot answer “show me how Clause 7.5.3 (control of documented information) works in your system,” it is not ready.
IATF 16949 — automotive
IATF 16949:2016 is the automotive Quality Management System standard that sits on top of ISO 9001:2015 with additional clauses for the automotive supply chain. A QMS used for automotive must handle Process FMEA, Control Plans, PPAP (Production Part Approval Process), MSA (Measurement System Analysis), and SPC (Statistical Process Control), and must accommodate customer-specific requirements (CSRs) from OEMs like General Motors, Stellantis, and Ford. Many IATF-certified suppliers also run layered process audits per AIAG CQI-8 as a CSR requirement; a QMS must run LPAs alongside formal internal audits with shared CAPA workflow.
For European automotive work, VDA 6.3 (the process audit standard published by the VDA QMC — the Quality Management Center of the Verband der Automobilindustrie, the German automotive industry association) is often layered on top of IATF. A QMS that supports automotive should support both audit methodologies natively.
AS9100 — aerospace and defense
AS9100 is the aerospace Quality Management System standard, also built on ISO 9001 with industry-specific additions for Key Characteristics, First Article Inspection (FAI), counterfeit-part prevention, configuration management, and product safety. QMS platforms used in aerospace must support FAI per AS9102, risk management per ISO 14971-equivalent practices, and stricter document control retention (often 10+ years). AS9100D is the current published revision; AS9100 Rev E has been in development with the IAQG and SAE. Readers should confirm the current revision status directly at sae.org and iaqg.org, as the development timeline is subject to change.
FDA cGMP and 21 CFR Part 11 — regulated manufacturing
For FDA-regulated manufacturers, the QMS must comply with the relevant Current Good Manufacturing Practice (cGMP) regime and 21 CFR Part 11 for electronic records and signatures. The cGMP regime differs by product type: pharmaceutical and biotech manufacturers comply with cGMP for finished pharmaceuticals at 21 CFR Parts 210 and 211 (unchanged by recent rulemaking); medical device manufacturers comply with the FDA Quality Management System Regulation (QMSR) at 21 CFR Part 820, which as of 2 February 2026 incorporates ISO 13485:2016 by reference (replacing the legacy Quality System Regulation framework).
Electronic records, signatures, and computer system assurance
Part 11 governs electronic records and signatures across both regimes; it requires secure audit trails, role-based access, electronic signatures bound to records, and the ability to produce records on demand for an FDA inspection. Computer system validation has traditionally been an IQ/OQ/PQ effort; FDA’s 2022 Computer Software Assurance (CSA) guidance now permits a risk-based approach that reduces this burden where appropriate.
For medical devices specifically, ISO 13485 is now the foundation of the FDA’s framework. As of 2 February 2026, the Quality Management System Regulation (QMSR) amended 21 CFR Part 820 to incorporate ISO 13485:2016 by reference, replacing the prescriptive QSR requirements. Part 820 as a regulatory location remains in force. The QMSR restructures the legacy Design History File, Device Master Record, and Device History Record under the ISO 13485 framework. Device manufacturers must maintain the equivalent records as required under ISO 13485 §§ 7.5.8, 8.2.6, and associated clauses, with FDA-specific requirements layered on top. Note that 21 CFR Part 820 as a regulatory location remains in force — it was amended, not repealed. Consultation with a regulatory affairs specialist is recommended for transition planning.
HACCP and GFSI standards — food safety
Food manufacturers run their QMS against HACCP (Hazard Analysis Critical Control Points) and a GFSI-recognized standard — SQF, BRCGS, FSSC 22000, or IFS. The QMS must support Critical Control Point monitoring, sanitation pre-operational inspections, allergen control, traceability one-up and one-down, and supplier approval for raw material. We cover this in depth elsewhere in our food safety + HACCP coverage.
How to choose QMS software — the buyer’s framework
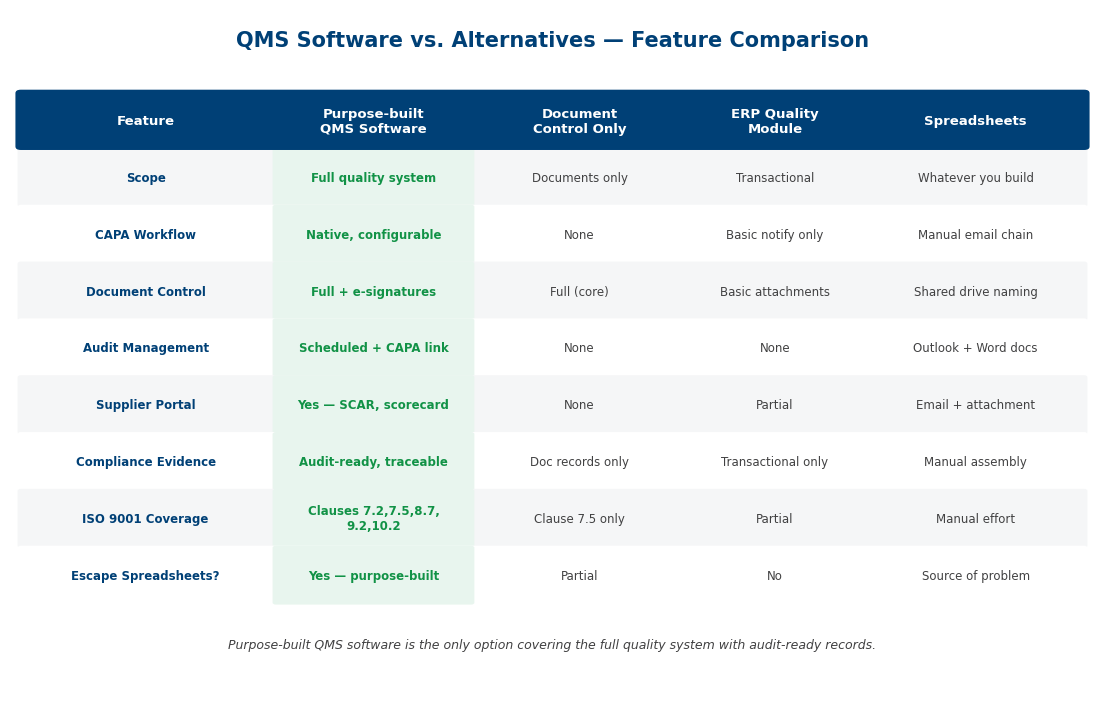
QMS selection failures are almost always traceable to one of five errors: buying a custom-coded platform that requires vendor consultants for every change, ignoring multi-site requirements, treating mobile as an afterthought, miscounting cloud-versus-on-premise TCO, or under-specifying the audit trail. The framework below is built to prevent each of them.
Configurable vs custom-coded workflows
The single biggest selection mistake is buying a platform that requires a vendor consultant to make any change. A control plan adds a parameter, a customer adds an inspection step, a regulator updates a clause — and every change becomes a change request, a quote, and a six-week delay. A configurable QMS lets the quality team itself adjust workflows, forms, dashboards, and approval routings without IT or vendor intervention.
Test for it in the demo: ask the vendor to add a question to an audit, change an approval routing, and create a new dashboard view in front of you. If they cannot or will not, the platform is custom-coded under the hood.
Multi-site vs single-site deployment
If you operate more than one site — and most QMS buyers do — the platform must handle multi-site natively. That means site-scoped data with cross-site visibility, site-specific document control with global master documents, site-specific KPIs with consolidated rollup, and role-based access that respects site boundaries. Many platforms claim multi-site but implement it as separate tenants with manual reconciliation. Verify it by asking to see one corporate dashboard with five live sites.
For global manufacturers, multi-language is the corollary requirement — auditors and operators need the platform in their working language, even when the controlled document is in English.
Mobile-first inspection vs office-only
The point of the QMS is to capture quality data at the point of work. Inspections done at a desk an hour after the shift, transcribed from paper, are unreliable and slow. A modern QMS runs natively on a tablet or phone, captures photo evidence at the question, works offline in connectivity-poor areas (welding bays, refrigerated rooms, customer sites), and syncs when the device reconnects. If the vendor’s demo is desktop-only, the platform is not mobile-first.
Cloud vs on-premise — TCO comparison
Cloud (SaaS) QMS is now the default. On-premise survives only in specific cases: classified defense work, certain validated pharma environments, or jurisdictions with strict data-residency rules. The cloud TCO advantage is significant — no infrastructure cost, automatic upgrades, faster validation in regulated environments because the vendor maintains the IQ/OQ baseline. For most manufacturers, the decision is which cloud, not whether cloud.
Data residency is the one question that requires explicit answer: where is the data hosted, can it be hosted in a specific region (EU, US, Canada, Australia), and what is the vendor’s posture on cross-border transfers? GDPR, CSDDD, and emerging US state privacy laws all create requirements here.
Audit trail and defensible evidence requirements
The QMS must produce an audit trail that holds up under scrutiny — not just from your internal team, but from a Notified Body certification audit, a customer audit, or a regulator. That means every record carries a timestamp, the user identity, the action taken, and (for electronic signatures) the credential used to sign. Records must be tamper-evident: changes are logged, not overwritten, and deleted records remain in the audit trail. This is what we mean by defensible evidence — proof that the action happened, by whom, when, and against what version of the controlled document.
A short checklist before issuing an RFP
- Can the quality team configure workflows, forms, and dashboards without the vendor?
- Does it run natively on mobile with offline capture and photo evidence?
- Does multi-site work as a true rollup, not separate tenants?
- Can CAPA enforce Verified Closure, not just signature closure?
- Does the audit trail capture timestamp, user, action, and document revision on every record?
- What is the realistic implementation timeline — and which customer reference can confirm it?
- What is the per-user, per-supplier, and per-site pricing model — and how does it scale at 2× and 5× volume?
A vendor that hedges on any of these is signalling a problem that will surface six months into your deployment.
See how Certainty supports your QMS
Configurable · Mobile-first · Multi-site · Verified Closure on every CAPA · One audit trail, every standard
See Certainty’s audit management softwareQMS implementation roadmap (6 steps)
A configurable QMS implementation does not need to be a 12-month change-management project. The six-step roadmap below is what we see succeed across mid-market manufacturers, completed in 8 to 16 weeks for a single-site rollout and 4–6 months for a multi-site enterprise.
Step 1 — Map current quality processes
Before any platform is configured, document how quality actually runs today. List every controlled document, every audit type and frequency, every non-conformance source, every training matrix, every supplier qualification step, every report your quality team produces. Mark which are mature (working as designed) and which are broken (paper, spreadsheet, “tribal knowledge”). The output is a one-page current-state map that becomes the implementation backlog.
This step is also where you discover redundancy — eight slightly different inspection checklists for the same process, three competing supplier scorecards, two parallel CAPA logs. Consolidation in step 1 saves months later.
Step 2 — Define document hierarchy and retention
Document control is the foundation. Define the hierarchy (Quality Manual → Procedures → Work Instructions → Forms → Records), the change-control workflow (draft → review → approve → train → release), the retention policy (typically 3 years minimum, 7+ for regulated industries, 10+ for aerospace and medical device), and the training-on-revision rule (who must re-acknowledge when a document changes).
Configure these in the platform before any inspections or audits are turned on. A document-control foundation that runs cleanly for two weeks gives the rest of the rollout a stable base.
Step 3 — Configure audit templates
Build the audit templates the business actually uses — internal audit against ISO 9001 or IATF 16949, layered process audits per CQI-8, supplier audits, food safety pre-op inspections, customer audit prep. Each template is anchored to a question library, an evidence requirement, and an escalation path. Keep templates short and observable — most failing audit programs have checklists that are too long and too subjective.
For multi-site rollouts, build the corporate-standard template first, then allow site-level variants for site-specific equipment or process. Resist the urge to let every site invent its own checklist.
Step 4 — Stand up CAPA workflow with Verified Closure
Configure the CAPA workflow as a closed loop: trigger → assignment → root-cause analysis → corrective action → preventive action → Verified Closure. Set the verification step as a separate gate, not an automatic closure. Specify which roles can verify (typically not the same person who performed the action), what evidence is required (follow-up audit, sample data, photographic), and what the verification window is (30, 60, or 90 days depending on action type).
This is the step most implementations skimp on. Spend two days here and you will save twelve months of repeat findings later.
Step 5 — Roll out training and go-live cadence
Train auditors, document owners, and CAPA owners on the platform — not just the how but the why. Train executives on the dashboards they will see during management review. Set a phased go-live: document control first (week 1), then audit management (week 3), then CAPA (week 5), then supplier quality (week 8). Each phase builds on the previous one.
Run a parallel period of 2–4 weeks where the new platform and the legacy paper/spreadsheet process operate in parallel. This catches gaps without breaking compliance during cutover.
Step 6 — Measure: COPQ, FPY, defect escape rate
Once the platform is live, measure. The leading indicators — audit completion rate, CAPA aging, training completion — tell you whether the system is being used. The lagging indicators — Cost of Poor Quality (COPQ), first-pass yield (FPY), defect escape rate, customer PPM — tell you whether quality is actually improving. Review monthly with the operations leadership team, quarterly with the executive team, annually as part of formal Management Review per ISO 9001 Clause 9.3.
We cover specific benchmarks in QMS KPIs and benchmarks below.
Defensible evidence and audit readiness
The job of a QMS is not to pass an audit — it is to make every day an audit-ready day. Defensible evidence is the discipline that gets you there: timestamped records, tamper-evident audit trails, user-attributable actions, and version-controlled documents that hold up under certification, customer, and regulatory scrutiny.
What auditors look for
ISO 19011:2026 is the international guideline for auditing management systems. It tells auditors — and tells you what auditors are trained to do. The four things every certification auditor and most customer auditors look for are:
- Evidence the process happened as documented. Records of inspections, training, calibration, CAPA — with timestamps that match the documented frequency.
- Traceability through the workflow. A finding traces to a CAPA traces to a root cause traces to a Verified Closure. Breaks in the chain are findings.
- Document and training alignment. Operators are trained on the current revision of the work instruction they are performing — not an obsolete version.
- Closed-loop improvement. Findings from prior audits have been actioned and have not recurred. Management Review (ISO 9001 Clause 9.3) shows continual improvement, not just compliance.
Audit trail requirements
A defensible audit trail captures, at minimum, on every record: who performed the action, what action, when (date and timestamp), against what version of the controlled document, with what evidence attached. Electronic signatures must be bound to the record per 21 CFR Part 11 for regulated industries, and per ISO 9001:2015 Clause 7.5.3 (control of documented information) for everyone else. Records must be tamper-evident — changes are logged, not silently overwritten.
The audit trail is also where you discover whether your platform is actually serving you. A QMS that allows records to be edited after closure, or signatures to be applied without authentication, will fail at customer audit even if it never fails an internal one.
Customer audit prep — automated evidence package
The most expensive QMS task in most quality teams’ calendar is preparing for a customer audit — pulling records, screenshotting dashboards, building evidence binders. A modern QMS automates this: when a customer audit is scheduled, the platform produces an evidence package for the scope (date range, processes audited, sites involved) including audit records, CAPA closure, training records, supplier evidence, and management review minutes. What used to take a quality engineer a week takes the platform an hour.

QMS KPIs and benchmarks

A QMS without KPIs is documentation infrastructure. A QMS with the right KPIs is a competitive advantage. Quality leaders run two sets of measures: leading indicators that predict whether the system is working, and lagging indicators that show whether quality is actually improving.
Leading indicators (measure program health)
| KPI | Definition | Mature-program benchmark |
|---|---|---|
| Audit completion rate | % of scheduled audits actually performed within the scheduled window | ≥ 95% |
| CAPA aging | Median days a CAPA stays open before Verified Closure | ≤ 30 days for routine; ≤ 10 days for safety-critical |
| Training completion on revision | % of role-affected operators retrained within 14 days of a document revision | ≥ 95% |
| Document overdue rate | % of controlled documents past their scheduled review date | ≤ 5% |
| Photo / evidence capture rate | % of audit questions completed with required evidence attached | ≥ 90% |
| Supplier scorecard freshness | % of active suppliers with a scorecard updated within the last quarter | ≥ 90% |
Leading indicators are the early warning system. If audit completion drops below 90%, the lagging indicators will follow 2–3 quarters later.
Lagging indicators (measure business outcomes)
| KPI | Definition | Mature-program benchmark |
|---|---|---|
| Cost of Poor Quality (COPQ) % of sales | Scrap + rework + warranty + inspection + field-failure cost as % of revenue | Mature programme target: below 5% of revenue when all cost categories are included. The 15–20% baseline figure is commonly cited from Juran and Crosby’s foundational quality cost research (1970s–1990s); current industry surveys suggest mature manufacturers typically achieve 2–5%. Organisations should establish their own baseline before benchmarking against published figures, as definitions and inclusions vary materially. |
| First-Pass Yield (FPY) | % of units produced right the first time, no rework | Trend should improve year over year |
| Defect escape rate | Customer-reported defects per million shipped (PPM) | Continuous downward trend |
| Customer complaint cycle time | Median days from customer complaint to Verified Closure | ≤ 30 days |
| Repeat finding rate | % of audit findings that recur within 12 months at the same location | ≤ 10% |
| Certification audit findings | Non-conformances raised in the most recent certification surveillance | Zero majors; trending down on minors |
The lagging indicators are where executive attention belongs. The leading indicators are where the quality team’s daily attention belongs. A QMS that surfaces both, drilled-down to source records, is the platform you want.
QMS across industries
The core QMS architecture is consistent across industries, but the regulatory overlay and operational specifics vary. Here is how it differs in the five largest industry segments — automotive, aerospace, food and beverage, medical device and pharma, and general industrial manufacturing.
Automotive (IATF 16949 / VDA 6.3)
Automotive quality is defined by the supply chain. A QMS in automotive must run Process FMEA, Control Plans, PPAP, MSA, SPC, and customer-specific requirements (CSRs) from each OEM. Layered process audits per CQI-8 are mandatory for GM and Stellantis suppliers. European OEMs frequently require VDA 6.3 process audits in addition. Multi-language matters — Tier 1 suppliers often run plants in Mexico, Eastern Europe, China, and India where the operator’s working language is not the controlled document’s language.
Aerospace (AS9100)
Aerospace adds traceability and counterfeit-part prevention to the QMS workload. First Article Inspection per AS9102 must be a first-class workflow. Key Characteristics flow from design through production with explicit measurement and inspection at every step. Document retention extends to 10+ years for flight-safety parts. The QMS audit-trail discipline is higher than in any non-medical industry.
Food and beverage (GFSI / HACCP)
Food QMS revolves around HACCP critical control points, sanitation pre-operational inspections, allergen control, and traceability one-up and one-down. The GFSI-recognized standards (SQF, BRCGS, FSSC 22000, IFS) each add their own audit scheme. A QMS for food must handle environmental monitoring (swab schedules, micro results), supplier approval for raw material, and a recall workflow that is testable on demand. We cover this in depth elsewhere in our food safety + HACCP coverage.
Medical device and pharma (FDA cGMP)
Regulated medical device manufacturers run their QMS against the FDA QMSR (21 CFR Part 820 as amended February 2026, incorporating ISO 13485:2016 by reference) and 21 CFR Part 11 for electronic records. Pharma runs under FDA cGMP at 21 CFR 210/211 (unchanged by the QMSR transition). Validation has traditionally been an IQ/OQ/PQ effort; FDA’s 2022 Computer Software Assurance (CSA) guidance now permits a risk-based approach in many cases. Design and development files (the QMSR replacement for the legacy Design History File terminology) are first-class objects. Risk management per ISO 14971 integrates with the CAPA workflow. eQMS is the right vocabulary here, and the implementation profile is heavier than non-regulated industries.
General industrial manufacturing (ISO 9001)
ISO 9001-only manufacturers — industrial equipment, fabrication, contract manufacturing — have the most flexibility in their QMS configuration. Document control, internal audit, CAPA, and supplier quality are the must-haves. Customer-specific requirements vary widely; the QMS must be configurable enough to handle a Tier 1 customer’s specific inspection package without a vendor change request.
Key Takeaways:
- A modern Quality Management Software (QMS) platform is the digital system of record for documents, audits, CAPA, training, supplier quality, and risk — replacing scattered spreadsheets and paper checklists with one defensible source of truth.
- The QMS capability spectrum spans document control, audit management, non-conformance and CAPA with Verified Closure, training, risk-based planning, multi-site reporting, and supplier quality — Certainty owns the audit-and-CAPA execution core and integrates with the document-control and training systems that complete the quality system.
- CAPA-forward QMS treats corrective and preventive action as the first-class workflow, not a back-office feature — and is the difference between a system that learns from findings and one that repeats them.
- Verified Closure is the discipline of treating “complete” and “effective” as two separate events; it is what certification auditors and customer auditors look for under ISO 9001 Clause 10.2 and IATF 16949 Clause 10.2.3.
- A QMS supports ISO 9001:2015, IATF 16949, AS9100, FDA cGMP / 21 CFR Part 11, HACCP / GFSI, and ISO 13485 in one configurable platform — industry-specific platforms are gradually losing share.
- The buyer’s framework: configurable workflows, multi-site native, mobile-first inspection, cloud TCO, audit trail with defensible evidence.
- A configurable QMS implementation runs 8–16 weeks per site, not the 6–18 months traditional eQMS deployments still quote.
- Measure leading indicators (audit completion, CAPA aging, training completion) and lagging indicators (COPQ, FPY, defect escape rate, repeat finding rate) — both, every month.
Frequently Asked Questions (FAQs)
What is the difference between QMS and eQMS?
QMS (Quality Management Software) is the umbrella category — any platform that runs quality processes (documents, audits, CAPA, training) digitally. eQMS (electronic Quality Management System) historically refers to heavyweight, validated platforms built for FDA-regulated industries with 21 CFR Part 11 compliance, IQ/OQ/PQ validation, and 6–18 month implementations. In practice, the terms are increasingly used interchangeably as configurable QMS platforms now support both regulated and non-regulated industries.
Is QMS software required for ISO 9001 certification?
No — ISO 9001:2015 does not require software. The standard requires a documented Quality Management System, documented information control (Clause 7.5), internal audits (Clause 9.2), non-conformance and CAPA (Clause 10.2), and management review (Clause 9.3). These can be implemented on paper. In practice, certification is significantly easier with software because the audit trail, version control, and traceability requirements are difficult to satisfy with spreadsheets alone at any meaningful scale.
Can QMS software handle multi-site and multi-language deployment?
Modern QMS software is built for multi-site multi-language operation. The platform should support site-scoped data with cross-site rollup, site-specific document variants with global master documents, operator interfaces in local working languages, and role-based access that respects site boundaries. Verify the multi-site claim by asking the vendor to demo one corporate dashboard with five live sites and one operator interface in two languages.
How long does QMS implementation take?
A configurable, cloud QMS for a single site typically goes live in 8 to 16 weeks: document control in weeks 1–3, audit management in weeks 4–6, CAPA in weeks 7–9, supplier quality in weeks 10–12, training and stabilization in weeks 13–16. Multi-site enterprise rollouts run 4–6 months. Traditional validated eQMS for regulated pharma or medical device runs 6–18 months because of validation (IQ/OQ/PQ) and change-control overhead.
How does QMS connect to ERP, MES, and SAP?
A modern QMS exposes a REST API and supports common integration patterns with ERP (SAP, Oracle, Microsoft Dynamics, NetSuite), MES (Rockwell, Siemens, Aveva), and BI tools (Power BI, Tableau, Looker). Typical integrations include pulling part numbers and bills of material from ERP into the QMS, pushing non-conformance and scrap data from the QMS back to ERP for cost accounting, syncing supplier master data, and feeding KPI dashboards from the QMS into corporate BI.
What is “CAPA-forward QMS”?
A CAPA-forward QMS is a quality management platform in which corrective and preventive action is the first-class workflow, not a back-office menu item. Every audit finding, non-conformance, customer complaint, and supplier issue routes directly into a CAPA with assigned owner, root-cause analysis, action evidence, and Verified Closure. The architecture inverts the older pattern in which CAPA was a feature inside the audit module — making CAPA the spine of the system instead of a side effect.
What is “Verified Closure”?
Verified Closure is the discipline of treating “complete” and “effective” as two separate events in a CAPA workflow. An action is complete when the assigned owner has performed it and provided evidence. A CAPA is closed only after a second verification — typically a follow-up audit, a sample inspection, or a fresh data point demonstrating the action prevented recurrence. Verified Closure is what auditors look for under ISO 9001 Clause 10.2 and IATF 16949 Clause 10.2.3.
How is QMS different from audit management software?
Audit management software is a focused tool for running audits and inspections — internal, supplier, layered process, food safety. QMS software is the umbrella platform that includes audit management plus document control, CAPA, training, supplier quality, and risk-based planning. Many organizations buy audit management software first as a fast-time-to-value starting point, then add CAPA, document control, and supplier quality on the same platform over time.
Is QMS software defensible evidence in a customer audit?
A properly implemented QMS produces defensible evidence — timestamped records, tamper-evident audit trails, user-attributable actions, version-controlled documents, and electronic signatures bound to records. The same defensibility that satisfies an ISO 9001 surveillance audit or an FDA inspection satisfies a customer audit. The test is whether the platform can reconstruct what happened, by whom, when, and against which document version for any record, on demand.
What does QMS software typically cost?
Mid-market configurable QMS platforms range from US$15,000 to US$200,000 annually depending on user count, site count, modules deployed, and supplier seats. Traditional validated eQMS for regulated industries can run $250,000 to $1M+ in year one including implementation and validation. The relevant cost comparison is total cost of ownership — platform fees plus implementation plus ongoing configuration plus the cost of not having a working QMS.
Related resources
Standards and guidance
- ISO 9001:2015 — Quality management systems — Requirements (ISO)
- ISO 13485:2016 — Medical devices QMS requirements (ISO)
- ISO 19011:2026 — Guidelines for auditing management systems (ISO)
- IATF 16949 — automotive sector QMS (IATF Global Oversight)
- FDA Quality Management System Regulation (QMSR) — Final Rule, effective 2 Feb 2026
- ASQ — Total Quality Management body of knowledge
Certainty platform resources
- CAPA Software: The Complete Guide
- EHS Software: The Complete Guide
- Layered Process Audits: The Complete Guide
- IATF 16949: A Practical Guide
- ISO 19011: A Practical Guide to Auditing Management Systems
- A Comprehensive Guide to Root Cause Analysis
- 8D Problem Solving: A Guide for Businesses
- DMAIC: A Practical Guide
- Audit Management Software
Ready to modernize your QMS?
Most QMS programs do not fail because the standards are unclear. They fail because evidence is scattered across spreadsheets and shared drives, CAPAs close without proof of effectiveness, and audit prep consumes weeks every cycle. Certainty consolidates the work onto one configurable platform that produces defensible evidence by design — and gives leadership real-time visibility across sites, suppliers, and product lines.


